O Sistema Sala Limpa é um padrão regulado de atividades e técnicas para o controle de áreas esterilizadas e ambientes controlados.
Tendo como definição de área Limpa, área esterilizada com controle ambiental definido em termos de fluxo de ar, pressão, temperatura. Além de umidade, ruído, vibração, iluminação, contaminação microbiana e por partículas. Projetada e utilizada de forma a reduzir a introdução, a geração e a retenção de contaminantes em seu interior.
Definição de Sala Limpa: Ambiente controlado utilizado para testes ou manufatura de produtos onde a contaminação por partículas presentes no ar interfere no resultado. Necessário em laboratórios químicos, laboratórios que produzem remédios, indústria de biotecnologia, indústria alimentícia, locais onde se manufaturam satélites espaciais, salas de operação, entre outros. É um local onde se consegue quantificar e mensurar o tamanho das partículas em suspensão. Sendo realizado com a existência de fluxo de ar turbulento e filtros absolutos HEPA, com eficiência é de 99,93% Fonte:(wikipédia)
Edificação: Conjunto de instalações arquiteturais que contém as áreas, instalações e recursos auxiliares.
Definição de Instalação: Espaço físico delimitado acrescido das máquinas, aparelhos, equipamentos e sistemas auxiliares utilizados para executar os processos.
Lote: Quantidade definida de matéria-prima, material de embalagem ou produto terminado fabricado em um único processo ou série de processos. Cuja característica essencial é a homogeneidade e qualidade dentro dos limites especificados. Na fabricação contínua, o lote corresponde a uma fração definida da produção. Algumas vezes é necessário dividir o lote em sub-lotes que posteriormente serão misturados para formar um lote homogêneo final.
Instalações para Sistema Sala Limpa
Instalações Físicas: As instalações devem ser localizadas, projetadas, construídas, adaptadas e mantidas de forma que sejam adequadas às operações a serem executadas. Seu projeto deve minimizar o risco de erros e possibilitar a limpeza e manutenção, de modo a evitar a contaminação cruzada. (contaminação de determinada matéria-prima, produto intermediário ou produto acabado com outra matéria-prima ou produto, durante o processo de manipulação). O acúmulo de poeira e sujeira ou qualquer efeito adverso que possa afetar a qualidade dos produtos.
.As instalações devem possuir ambientes que quando considerados em conjunto com as medidas destinadas a proteger as operações de fabricação, apresentem dessa forma, risco mínimo de contaminação dos materiais ou produtos neles manipulados.
.As instalações utilizadas na fabricação de medicamentos devem ser projetadas e construídas de forma a possibilitar a limpeza adequada.
.Devem ser mantidas em bom estado de conservação, higiene e limpeza. Precisa ser assegurado que as operações de manutenção e reparo não representem qualquer risco à qualidade dos produtos.
 |
| Instalações Sala Limpa |
As Áreas Internas
.As superfícies interiores (paredes, piso e teto) necessitam ser revestidas de material liso, impermeável lavável e resistente. Livres de juntas e rachaduras, de fácil limpeza, permitindo a desinfecção e não devendo liberar partículas.
.O fornecimento de energia elétrica, iluminação, ar acondicionado (temperatura e umidade) e ventilação, devem ser apropriados. De modo a não afetar direta ou indiretamente, os produtos durante os processos de fabricação e todavia, o armazenamento ou o funcionamento adequado dos equipamentos
.As tubulações, luminárias, pontos de ventilação e outras instalações serão projetadas e instaladas de modo a facilitar a limpeza. Sempre que possível o acesso para manutenção deve estar localizado externamente as áreas de produção.
.As tubulações fixas destinadas à condução de fluídos, devem ser devidamente identificadas, conforme legislação vigente e quando aplicável, a direção do fluxo deve ser indicada. Quando se tratar de gases e líquidos perigosos, devem ser empregados conexões.
.As instalações devem ser projetadas e equipadas de forma a permitirem a máxima proteção contra a entrada de insetos e outros animais.
Fonte: Resolução RDC nº 134, de 13 de julho de 2001, revogada pela Resolução RDC nº 210, de 04 de agosto de 2003.
Classificação para Sistema Sala Limpa
Para garantir a devida proteção ao ser humano, aos processos e aos produtos, a pureza de ar exigida em uma sala limpa deve ser determinada conforme as necessidades de cada situação específica. Afim de distinguir sistematicamente diferentes níveis de qualidade de áreas limpas, foram estabelecidas classes de pureza de ar.
As salas limpas são classificadas através das Normas, em função da pureza de seu ar interior, ou seja, da concentração de partículas por unidade de volume de ar.
A classificação de salas limpas contida na parte 1 da ISO 14644 é a classificação mundialmente mais utilizada atualmente, nela as Salas Limpas estão classificadas em nove classes. Podemos comparar essa nova norma técnica com as antigas normas US. FED STD 209D e US. FED STD 209E (que estão caindo em desuso mas que ainda são muito conhecidas dos especialistas):
ISO 14644-1 US. FED STD 209D US. FED STD 209E
 |
| Calassificação Sala Limpa |
As Salas Limpas Classe 1 a Classe 5 funcionam sob regime de fluxo de ar laminar e tem as seguintes principais características:
- A reposição total do ar acontece a cada seis segundos
- O ar flui uniformemente a partir do forro a uma taxa de 0,45 metros por segundo
- Há filtros por todo o forro
- Não há correntes de ar dentro do ambiente controlado
- Há um fluxo de ar uniforme pelo ambiente sem dispersão transversal de partículas
- O ar é retirado do ambiente somente por exaustão pelo piso.
Salas Limpas Classe 6 a Classe 9 funcionam sob regime de fluxo de ar turbulento e tem as seguintes principais características:
- O ar flui a partir de difusores no forro a uma taxa de 0,45 metros por segundo
- Há correntes de ar dentro do ambiente controlado
- Permite-se a existência de dispersão transversal de partículas dentro do ambiente controlado
- O ar é retirado do ambiente por exaustão pelo piso ou pelo forro.
Instrução
Normativa – Classificação
Instrução
Normativa - Classificação do Ar e Equipamentos - Salas Limpas
A
Instrução Normativa 35/2019 especifica a classificação das áreas limpas e dos
equipamentos que fornecem ar limpo ao sistema; e dispõe sobre as Boas Práticas
de Fabricação complementares a Medicamentos Estéreis.
As salas
limpas e os equipamentos que fornecem ar limpo devem ser assim, classificados
de acordo com a versão vigente da norma ISO 14644-1; seguindo os métodos
de ensaio da ISO 14644-3.
A
classificação deve
claramente distinguir-se do monitoramento ambiental das operações em processo.
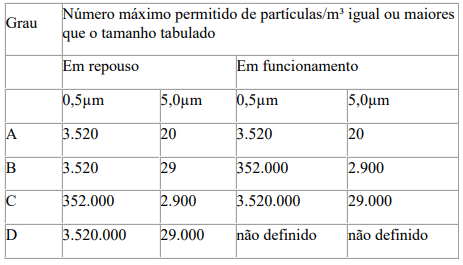
A
concentração máxima permitida de partículas no ar para cada grau assim, dada na
tabela abaixo.
 |
| Calassificação Sala Limpa |
Para fins
de classificação em zonas Grau A, um volume mínimo de 1m³ deve ser amostrado
por ponto de amostragem.
Para o Grau
A; a classificação para partículas ≥0,5μm é ISO Classe 5 e para partículas
≥5,0 μm é ISO M(20; 5μm); LSAPC, tanto em repouso como em operação.
Para o Grau
B; a classificação para partículas no ar ≥ 0,5μm é ISO Classe 5 em repouso
e ISO Classe 7 em operação, para partículas ≥ 5,0 μm é ISO M(29; 5μm); LSAPC em
repouso e ISO Classe 7 em operação.
Para o Grau
C; a classificação de partículas no ar é ISO Classe 7 em repouso e ISO
Classe 8 em operação, para ambos os tamanhos de partículas considerados no art.
10.
Para o Grau
D; a classificação de partículas no ar é ISO Classe 8 em repouso, para
ambos os tamanhos de partículas considerados no art. 10.
Para fins de classificação
Para fins
de classificação, as normas que determinam os métodos e procedimento de ensaios
são a ISO 14644-1-2015 (contagem de partículas) e; ISO 14644-3 FDIS –
2019 para todos os demais ensaios pertinentes.
Os
contadores de partículas portáteis com comprimento curto da tubulação de
amostragem devem ser assim, usados para fins de classificação; devido à taxa
relativamente mais alta de precipitação de partículas ≥5.0μm em sistemas
remotos com longos comprimentos de tubulação de amostragem.
Amostradores
isocinéticos de ar devem ser assim, usados para as áreas limpas com fluxo de ar
unidirecional.
A
classificação “Em operação” pode ser assim, demonstrada durante as operações de
fabricação operações simuladas; ou dessa forma, durante as simulações de
processo asséptico, visto que o pior cenário é requerido durante estas.
A norma ISO
14644-2 provê informações sobre os testes para demonstração da conformidade
contínua com o grau de limpeza estabelecido.
Fonte:
antigo.anvisa.gov.br/documents
HVAC - Ar Condicionado Sistema Sala Limpa
Sistema de Ar e Ventilação: As áreas de produção devem possuir sistema de ventilação efetivo, com unidades de controle de ar incluindo o controle de temperatura e, quando necessário, de umidade e filtração apropriados aos produtos nela manipulados, às operações realizadas e às condições do ambiente. Essas áreas devem ser regularmente monitoradas durante o período de produção e em repouso, a fim de assegurar o cumprimento das especificações da área.
As áreas limpas devem ter um sistema de ventilação que insufle ar filtrado e mantenha uma pressão positiva da área em relação às zonas circundantes. A ventilação deve ser eficiente e adequada às condições exigidas. Especial atenção deve ser dada as zonas de maior risco, onde o ar filtrado entra em contato com os produtos e os componentes limpos.
 |
| Grelha de insuflamento Sistema sala limpa |
Pode ser necessário que as diversas recomendações relativas ao suprimento de ar e aos diferenciais de pressão sejam modificadas no caso de ser necessário a contenção de materiais patogênicos, altamente tóxicos, radioativos ou materiais com vírus vivos ou bacterianos.
Em alguns processos, pode ser necessária a utilização de instalações destinadas a descontaminação e ao tratamento do ar que estiver saindo da área limpa. |
| Grelha de retorno Sistema sala limpa |
Deve ser demonstrado que o sistema de ar não constitui risco de contaminação. Deve ser assegurado que o mesmo não permita a disseminação de partículas originadas das pessoas, equipamentos ou operações, para as zonas de produção de maior risco. Um sistema de alarme deve ser instalado para indicar a ocorrência de falhas no sistema de ventilação. Além disso, deve ser colocado um indicador de diferencial de pressão entre as áreas onde tal diferença for importante. As diferenças de pressão devem ser registradas.
Calibração para Sistema Sala Limpa
Calibração: Conjunto de operações que estabelece, sob condições especificadas, a relação entre os valores indicados por um instrumento ou sistema de medição ou valores representados por uma medida materializada ou um material de referência, e os valores correspondentes
Os equipamentos e instrumentos utilizados nos procedimentos de medidas, pesagens, registros e controles devem ser submetidos a manutenção e a calibração a intervalos pré-estabelecidos e os registros de tais operações devem ser mantidos. Para assegurar um funcionamento satisfatório, os instrumentos devem ser verificados diariamente ou antes de serem utilizados para ensaios analíticos. As datas de calibração, manutenção e de quando devem ser feitas as futuras calibrações, devem estar claramente estabelecidas e registradas.
A Qualificação para Sistema Sala Limpa
Operações documentadas de acordo com um plano de testes pré-determinados e critérios de aceitação definidos, garantindo que componentes, equipamentos e instalações estejam adequados ao uso pretendido.
Qualificação de equipamentos (QE) Conjunto de operações que estabelece sob condições especificadas, que os resultados dos testes de determinado equipamento demonstram que o mesmo apresenta o desempenho previsto. Os instrumentos e sistemas de medição devem estar calibrados.
Qualificação de instalação (QI) Conjunto de operações que estabelece, sob condições especificadas, que a instalação dos equipamentos, utilidades, instrumentos de pesagem e medidas e áreas de produção; na fabricação de medicamentos, foram selecionados adequadamente e encontram-se corretamente instalados, de acordo com as especificações estabelecidas.
Qualificação operacional (QO) Conjunto de operações que estabelece, sob condições especificadas, que o sistema ou sub-sistema apresenta desempenho conforme previsto, em todas as faixas operacionais consideradas. Todos os equipamentos utilizados na execução dos testes, devem ser identificados e calibrados antes de serem usados.
Validação para Sistema Sala Limpa
Validação: Ato documentado que atesta que qualquer procedimento, processo, equipamento, operação ou sistema realmente conduza aos resultados esperados. Os estudos de validação constituem parte essencial das BPF e devem, portanto ser conduzidos de acordo com protocolos prédefinidos. Deve ser mantido relatório escrito com o resumo dos resultados obtidos e as conclusões. Os processos e procedimentos devem ser estabelecidos, de acordo com os resultados do estudo de validação e devem sofrer revalidações periódicas, para que seja assegurado que os mesmos permaneçam capazes de atingir os resultados planejados.
Atenção especial deve ser dada à validação dos processos, dos ensaios de controle e dos procedimentos de limpeza. Os processos considerados críticos devem ser validados, concorrente, prospectiva e/ou retrospectivamente.
Os processos de validação requerem a colaboração mútua de todos os setores envolvidos tais como: desenvolvimento, produção, engenharia, manutenção, garantia da qualidade e controle de qualidade.
A validação permite aperfeiçoar os conhecimentos dos processos produtivos e desta forma assegurar que os processos encontram-se sob controle. Diminuir os riscos de desvio de qualidade. Diminuir os riscos da não conformidade aos requisitos estabelecidos. Reduzir a quantidade de testes de controle de qualidade nas etapas de controle em processo e no produto terminado.
Tipos de validação de processo Sistema Sala Limpa
Validação prospectiva: A validação prospectiva é um ato documentado, baseado na execução de um plano de testes previamente definidos, que demonstre que um novo sistema, processo, equipamento ou instrumento, ainda não operacionalizado, satisfaz as especificações funcionais e expectativas de desempenho.
A validação prospectiva é realizada durante o estágio de desenvolvimento do produto, através da análise dos riscos do processo de fabricação, o qual é detalhado em passos individuais; estes, por sua vez, são definidos com base na experiência passada para determinar se os mesmos podem ocasionar situações críticas.
Devem ser identificados os pontos críticos, avaliados quanto a sua probabilidade e extensão, e suas causas pesquisadas.
Os planos de pesquisa, são definidos, estabelecendo as prioridades e sua avaliação final.
Se, ao final do processo de validação, os resultados são aceitáveis, o processo é satisfatório. Se os resultados forem insatisfatórios deve-se buscar modificação no processo até que o mesmo apresente resultados aceitáveis. Esta forma de validação é essencial para limitar o risco de erros que ocorrem em escala de produção industrial.
Validação concorrente ou simultânea: A validação concorrente é realizada durante a produção de rotina. Este método somente é eficaz caso no estágio de desenvolvimento do produto tenha resultado no conhecimento adequado das bases do processo. Os primeiros lotes de produção industrial devem ser monitorados da forma mais abrangente possível. A natureza e as especificações dos testes subseqüentes em processo e finais estão baseados na avaliação dos resultados do referido monitoramento.
A validação concorrente, junto com uma análise de tendência incluindo os estudos de estabilidade, deve ser realizada com a extensão adequada ao longo da vida do produto.
Validação de processo Retrospectiva - Sistema Sala Limpa
Validação retrospectiva: Validação retrospectiva é um ato documentado, baseado na revisão e análise de registros históricos, atestando que um sistema, processo, equipamento ou instrumento, já em uso, satisfaz as especificações funcionais e expectativas de desempenho.
A validação retrospectiva envolve a verificação da experiência passada de produção, assumindo-se que a composição, procedimentos e equipamentos permanecem inalterados; a referida experiência e os resultados dos testes de controle em processo e final são avaliados. As dificuldades e defeitos registrados na produção são analisados para determinar os limites dos parâmetros do processo. Pode ser realizada uma análise de tendência para determinar a extensão na qual os parâmetros do processo encontram-se dentro da faixa permissível.
Obviamente, a qualificação retrospectiva não é uma medição da garantia da qualidade em si própria, e nunca deve ser aplicada a novos processos ou produtos. Somente pode ser considerada em circunstâncias especiais, p. ex., quando os requisitos de validação são estabelecidos pela primeira vez dentro da empresa.
Neste caso a validação retrospectiva pode ser útil para estabelecer as prioridades do programa de validação. Caso os resultados da validação retrospectiva sejam positivos, isto indica que o processo não tem necessidade de atenção imediata e pode ser validado de acordo com a programação normal.
Normas Técnicas para Sistema Sala Limpa
Normas Técnicas da ABNT: Clicando neste link, você será levado ao site da ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS e poderá fazer sua pesquisa por palavra, código, tema ou data.
Consulte diretamente as normas a seguir, relacionadas ao tema Sala Limpa (área limpa).
Normas vigentes: As salas limpas possuem suas normas específicas que definem, entre outros aspectos, a classificação dos ambientes quanto aos níveis de contaminação, técnicas construtivas adequadas, procedimentos operacionais e procedimentos de teste para verificação de limpeza do ar interior.
Federal Standard 209 – Airborne Particulate Cleanliness Classes in Clean Rooms and Clean Zones
A primeira edição da Norma Federal Standard 209 data de 1963, nos Estados Unidos, sob o título “Clean Room and Workstation Requirements, Controlled Environments”. A Norma, já obsoleta, define classes de limpeza do ar e estabelece métodos para medição da limpeza do ar em ambientes controlados.
ISO 14644 - Cleanrooms and associated controlled environments Esta norma ISO foi primeiramente aplicada na União Européia em 1999 e em seguida nos Estados Unidos em 2001, onde vêm tomando o lugar da Federal Standard 209. A ISO 14644 é composta por 8 partes:
ABNT/CB-46 Áreas Limpas e Controladas
NBRISO14644-1- parte 1: Classificação da limpeza do ar- Abrange a classificação da limpeza do ar em salas limpas e ambientes controlados associados, exclusivamente em termos de concentração de partículas em suspensão no ar. Para o objetivo da classificação, são consideradas somente as populações de partículas com distribuições cumulativas baseadas em tamanhos limiares (limite inferior) variando entre 0,1 micrometro e 5 micrometros.
NBRISO14644-2- Parte 2: Especificações para ensaios e monitoramento para comprovar a contínua conformidade com a ABNT NBR ISO 14644-1- Especifica requisitos para ensaios periódicos para sala ou zona limpa, para comprovar a contínua conformidade com a ABNT NBR ISO 14644-1, para classe designada de limpeza do ar para partículas em suspensão.
NBRISO14644-3 (Inglês)- Part 3: Metrologia e métodos de teste - Descreve os métodos de teste de salas limpas para validar sua condição de conformidade com a Norma.
NBRISO14644-4- Salas limpas e ambientes controlados associados - Parte 4: Projeto, construção e partida- Especifica os requisitos para o projeto e construção de instalações de salas limpas, porém não prescreve meios tecnológicos ou contratuais específicos para atender à conformidade com esses requisitos. Destina-se a usuários, fornecedores e projetistas de instalações de salas limpas e fornece uma lista de verificação para os parâmetros importantes de desempenho. Proporciona um guia de construção, incluindo requisitos para partida e qualificação. Os elementos básicos de projeto e construção, necessários para assegurar uma operação satisfatória contínua, são identificados levando-se em consideração os aspectos relevantes de operação e manutenção.
Operações em Salas Limpas
NBRISO14644-5- Parte 5: Operações- Especifica requisitos básicos para as operações em salas limpas. Tem por objetivo atender às pessoas que utilizam e operam uma sala limpa. Os aspectos de segurança não ligados diretamente ao controle da contaminação não são levados em consideração nesta parte da ABNT NBR ISO 14644 e regulamentos de segurança nacionais e locais devem ser respeitados. Considera todas as classes de salas limpas utilizadas para a produção de todos os tipos de produtos. Em conseqüência, tem grande abrangência na sua aplicação e não leva em conta as exigências específicas de indústrias em particular.
NBRISO14644-6 (Inglês)- Part 6 – Termos e definições – Fornece uma coleção de termos e definições sobre salas limpas.
NBRISO14644-7- Parte 7: Dispositivos de separação (compartimentos de ar limpo, gloveboxes, isoladores, miniambientes)- Especifica os requisitos mínimos de projeto, construção, instalação, execução de ensaios e aprovação dos dispositivos de separação, nos aspectos onde eles diferem das salas limpas.
NBRISO14644-8 - (Inglês)-Part 8 – Contaminação molecular -Aborda a contaminação gasosa em salas limpas.
ISO14698 (Inglês)- Cleanrooms and associated controlled environments – Biocontamination control . Esta norma ISO é composta de três partes:
ISO/DIS 14698-1 Part 1 – Princípios gerais e métodos – Aborda os métodos de medição de microorganismos em salas limpas.
ISO/DIS 14698-2 Part 2 – Avaliação e interpretação de dados – Fornece informações de como tratar as informações obtidas das medições de microorganismos em salas limpas.
ISO/DTR 14698-3 (este original está disponível como um relatório técnico do esboço)- Part 3 – Metodologia para medição da eficiência de processos de limpeza – Determina a eficiência de processos de limpeza utilizados em salas limpas.
Classificação e controle de contaminação
NBR13700- Classificação e controle de contaminação- Estabelece classes-padrão de limpeza do ar e provê classes intermediárias para salas e zonas de trabalho limpas, baseadas em concentrações especificadas de partículas em suspensão no ar. Prescreve métodos para verificação da classe e requer um plano estabelecido para monitoramento de limpeza do ar. Também fornece um método para determinação e descrição das concentrações (indicador U) de partículas ultrafinas.
NBR15137- Sistemas espaciais - Controle de limpeza e de contaminação- Estabelece os requisitos gerais para o controle de limpeza e de contaminação a ser aplicado pela cadeia de fornecedores para o desenvolvimento de sistemas espaciais, incluindo as instalações de processamento de solo, equipamentos de apoio no solo, veículos lançadores, cargas úteis e operações na órbita e processamento em solo. Também fornece as diretrizes para o estabelecimento de um programa de controle de limpeza e de contaminação.
Consulte outros canais:
Normas Técnicas IMETRO- A Biblioteca Online do Inmetro tem como objetivo facilitar o acesso, o uso e a disseminação do seu acervo especializado aos clientes internos e externos, contribuindo para atender as necessidades de informação. Os usuários poderão ter acesso às Referências Bibliográficas de todo o acervo especializado do Inmetro, conhecer as novas aquisições e sua disponibilidade para consulta.
SBCC - Sociedade Brasileira de Controle de Contaminação Procure por firmas que vendem pacotes prontos para a montagem desse tipo de sala.
Normas Legislativas para Sistema Sala Limpa
Normas Legislativas ANVISA, (Legilação em vigilância sanitária), Site do Ministério da Saúde/Agência Nacional de Vigilância Sanitária veja todas as leis vigentes do setor , em especial sala limpa (área limpa) recomendamos que a pesquisa seja feita de trás para frente, pois a classificação é feita por data.
Glossário ANVISA- Pesquisar termos e Conceitos- Os termos constantes neste glossário foram identificados e conceituados dentro das normas legais. Assim a utilização destes termos, nos mecanismos de busca das bases de dados de texto completo, alem de facilitar a pesquisa agrupa legislações relacionadas buscando apresentar atos jurídicos que tenham no seu conteúdo o termo usado na pesquisa facilitando a localização do ato de interesse do usuário.
Resoluções Relacionadas
Resolução RDC nº 352, de 23 de dezembro de 2002 -Dispõe sobre o Regulamento Técnico de Boas Práticas de Fabricação para Estabelecimentos Produtores/Industrializadores de Frutas e ou Hortaliças em Conserva e a Lista de Verificação das Boas Práticas de Fabricação para Estabelecimentos Produtores/ Industrializadores de Frutas e ou Hortaliças em Conserva.
Resolução RDC nº 275, de 21 de outubro de 2002 -Dispõe sobre o Regulamento Técnico de Procedimentos Operacionais Padronizados aplicados aos Estabelecimentos Produtores/Industrializadores de Alimentos e a Lista de Verificação das Boas Práticas de Fabricação em Estabelecimentos Produtores/Industrializadores de Alimentos.
A Resolução RDC nº 267, de 25 de setembro de 2003 -Dispõe sobre o Regulamento Técnico de Boas Práticas de Fabricação para Estabelecimentos Industrializadores de Gelados Comestíveis e a Lista de Verificação das Boas Práticas de Fabricação para Estabelecimentos Industrializadores de Gelados Comestíveis.
Resolução RDC nº 210, de 04 de agosto de 2003 -Determina a todos os estabelecimentos fabricantes de medicamentos, o cumprimento das diretrizes estabelecidas no Regulamento Técnico das Boas Práticas para a Fabricação de Medicamentos, conforme ao Anexo I da presente Resolução.
A Resolução RDC nº 249, de 13 de setembro de 2005 -Determina a todos os estabelecimentos fabricantes de produtos intermediários e de insumos farmacêuticos ativos, o cumprimento das diretrizes estabelecidas no REGULAMENTO TÉCNICO DAS BOAS PRÁTICAS DE FABRICAÇÃO DE PRODUTOS INTERMEDIÁRIOS E INSUMOS FARMACÊUTICOS ATIVOS, conforme anexo I da presente Resolução.
Exigências para o Funcionamento dos Estabelecimentos
Resolução nº 169, de 19 de junho de 1996 -Norma Técnica que disciplina as exigências para o funcionamento dos estabelecimentos que realizam procedimentos médico-círúrgicos ambulatoriais, no âmbito do Estado de São Paulo. ítens 9.2.8 a 9.2.8.2
Lei nº 10813, de 24 de maio de 2001 -Dispõe sobre a proibição de importação, extração, beneficiamento, comercialização, fabricação e a instalação, no Estado de São Paulo, de produtos ou materiais contendo qualquer tipo de amianto.
Lei nº 7802, de 11 de julho de 1989 -Dispõe sobre a pesquisa, a experimentação, a produção, a embalagem e rotulagem, o transporte, armazenamento, a comercialização, a propaganda comercial, a utilização, a importação, a exportação, o destino final dos resíduos da embalagens, o registro, a classificação, o controle, a inspeção e a fiscalização de agrotóxicos, seus componentes e afins, e dá outras providências.
A Lei nº 6938, de 31 de agosto de 1981 -Dispõe sobre a Política Nacional do Meio Ambiente, seus fins e mecanismos de formulação e aplicação, e dá outras providências.
Resolução RDC nº 73, de 13 de abril de 2004 -Fascículo 5 da Parte II da 4ª Edição da Farmacopéia Brasileira
Esterilização
e Assepsia
Esterilização
e Assepsia, a Instrução Normativa 35/2019 especifica os processos de produção
dos medicamentos esterilizados terminalmente e medicamentos preparados
assepticamente.
- A preparação dos materiais e
da maioria dos medicamentos deve ser assim, feita em ambiente de no mínimo
Grau D e; dessa forma, a fim de oferecer um baixo risco de
contaminação microbiológica e de partículas, que seja adequado a filtração
e esterilização.
- Em situação de risco alto ou
incomum de contaminação microbiológica para o medicamento; por exemplo,
quando o produto suporta ativamente o crescimento microbiológico; quando deve ser
assim, mantido por um longo tempo antes da esterilização ou ainda mais,
quando o produto é; necessariamente, não processado em
tanques fechados; a preparação deve ser assim, realizada em
ambiente de Grau C.
- O envase de medicamentos
esterilizados terminalmente deve ser assim, realizado minimamente em área Grau
C.
- Em situação de risco incomum
de contaminação pelo ambiente; por exemplo, quando o processo de
envase é lento, quando os recipientes são de boca larga;
ainda mais quando estão necessariamente expostos por mais de alguns
segundos antes da selagem; o envase deve ser assim, realizado
em área Grau A com, minimamente Grau C no ambiente circundante.
- A preparação e o envase de
pomadas, cremes, suspensões e emulsões deve ser assim, normalmente
realizado em uma área Grau C, antes de sua esterilização terminal.
- A Instrução Normativa 35/2019
especifica os processos de produção dos medicamentos preparados
assepticamente.
- Os materiais, após a
lavagem, devem ser assim, manuseados minimamente em um ambiente de Grau
D.
O manuseio
- O manuseio de materiais e
matérias-primas estéreis; a menos que submetidos à esterilização
ou filtração através de um filtro de retenção de microorganismos;
posteriormente no processo deve ser assim, realizado em área
Grau A com uma área Grau B circundante.
- A preparação de soluções que
serão esterilizadas por filtração durante o processo deve ser assim,
realizada em uma área Grau C.
- Caso as soluções não sejam
filtradas, a preparação dos materiais e medicamentos deve ser assim; realizada em uma
área Grau A e da mesma forma, circundada por
Grau B.
- O manuseio e o envase de
produtos preparados assepticamente deve ser assim, feito em uma área Grau
A e; com Grau B como área circundante.
- Antes do término da
colocação das tampas; a transferência de recipientes parcialmente
fechados; como os utilizados em liofilização; deve ser assim, feita em
área Grau A com Grau B circundante ou em bandejas de
transferência seladas em um ambiente Grau B.
- A preparação e o envase de
pomadas, cremes, suspensões e emulsões estéreis; deve ser assim, feita em
uma área de Grau A, circundada por Grau B e; quando o
medicamento estiver exposto e não for posteriormente filtrado.
Fonte:
antigo.anvisa.gov.br/documents
Fabricação
de Medicamentos Estéreis
Fabricação
de Medicamentos Estéreis, Instrução Normativa nº 35, de 21 de agosto de 2019.
De acordo
com o Ministério da Saúde - (MS); e a Agência Nacional de Vigilância Sanitária
– (ANVISA); A Instrução Normativa nº 35, de 21 de agosto de 2019; publicada no
Diário Oficial da União nº 162, de 22 de agosto de 2019; dispõe sobre as Boas
Práticas de Fabricação complementares a Medicamentos Estéreis.
- Esta Instrução Normativa
possui o objetivo de adotar as diretrizes de Boas Práticas de Fabricação
de Medicamentos Estéreis do Esquema de Cooperação em Inspeção
Farmacêutica, PIC/S; como requisitos complementares a serem assim,
seguidos na fabricação de medicamentos estéreis; em adição às Diretrizes
Gerais de Boas Práticas de Fabricação de Medicamentos; e se aplica
às empresas que realizam assim, as operações envolvidas na fabricação de
medicamentos estéreis, incluindo os medicamentos experimentais.
As operações
- De modo geral, a fabricação
de medicamentos estéreis deve ser assim, realizada em áreas limpas; onde a
entrada seja assim, efetuada por antecâmaras para pessoal e/ou
equipamentos e materiais.
- As áreas limpas devem ser
assim, mantidas em um apropriado padrão de limpeza e receber ar que tenha
passado por filtros de eficiência apropriada.
- As várias operações de
preparação de materiais, preparação do medicamento e envase devem ser
assim, realizadas em áreas separadas dentro das áreas limpas.
- As operações de fabricação
são assim, divididas em duas categorias, sendo: primeiro, aquelas onde o
produto passa assim, por esterilização terminal; e segundo, aquelas que
são conduzidas assepticamente em alguma ou em todas as etapas.
- As áreas limpas para a
fabricação de medicamentos estéreis são assim, classificadas, de acordo
com as características exigidas do ambiente.
- Cada operação de fabricação
requer um nível de limpeza ambiental adequado no estado operacional; a fim
de minimizar os riscos de contaminação do medicamento ou dos materiais que
estão sendo trabalhados por material particulado ou microbiológico.
- Para atender às condições
“em operação”, as áreas limpas devem ser assim; projetadas para atingir
certos níveis especificados de limpeza do ar no estado “em repouso”.
- O estado “em repouso” é a
condição em que a instalação está montada e em funcionamento; com todos os
equipamentos de produção, mas sem pessoal presente.
- O estado “em operação” é a
condição em que a instalação está funcionando; em um modo de operação
definido com um número especificado de funcionários trabalhando.
- Os estados “em operação” e
“em repouso” devem estar definidos para cada sala limpa ou conjunto de salas limpas.
Na fabricação de medicamentos estéreis quatro graus
de limpeza podem ser assim, distinguidos:
I -Grau A; A zona para as operações de
alto risco como, por exemplo, a zona de envase; onde estão os reservatórios de
tampas, ampolas abertas e frascos-ampolas e onde são feitas conexões
assépticas. Normalmente, essas condições são assim, fornecidas por uma estação
de trabalho com fluxo de ar unidirecional ou isolador. Os sistemas de fluxo de
ar unidirecional devem assim, fornecer uma velocidade de ar homogênea na faixa
de 0,36 a 0,54m/s (valor de referência); medida na posição de trabalho das
estações de trabalho com fluxo de ar unidirecional abertas. A manutenção do
padrão de fluxo de ar unidirecional deve ser assim, demonstrada e validada. Um
fluxo de ar unidirecional e com velocidades mais baixas pode ser assim, usado
em isoladores e caixas com luva.
II -Grau
B; O
ambiente circundante da área Grau A, ou seja, a zona que circunda as
preparações e o envase assépticos.
III
-Graus C e D; Áreas
limpas para a realização de etapas menos críticas da fabricação de medicamentos
estéreis.
Fonte:
antigo.anvisa.gov.br/documents
Tecnologias
e Processos
Tecnologias
e Processos, especificação das tecnologias de Isoladores, Sopro,
Envase e Selagem nas Salas Limpas.
- A Instrução Normativa
35/2019 especifica a utilização da tecnologia de isoladores para minimizar
as intervenções humanas; em áreas de processamento pode resultar em uma
diminuição significativa no risco de contaminação microbiológica; dos
produtos preparados assepticamente a partir do ambiente.
- Existem muitos desenhos
possíveis de isoladores e dispositivos de transferência.
- O isolador e o ambiente
circundante devem ser assim, projetados de modo que a qualidade do ar;
requerida para as respectivas áreas, possa então, ser alcançada.
- Os isoladores são
construídos com materiais mais ou menos propensos a perfurações e vazamentos.
- Os dispositivos de
transferência podem variar de uma única porta a projetos de dupla porta;
ou até sistemas totalmente selados, que incorporam assim, mecanismos de
esterilização.
- A transferência de materiais
para dentro e para fora da unidade é em suma, uma das maiores fontes
potenciais de contaminação.
- A área dentro do isolador é
a área para operações de alto risco; embora seja reconhecido que um fluxo
de ar unidirecional possa assim, não existir na posição de trabalho destes
dispositivos.
- A classificação do ar
requerida para o ambiente circundante depende do desenho do isolador e de
sua aplicação.
- O ambiente circundante deve
ser controlado e, para o
processamento asséptico, deve haver uma classificação de no mínimo grau D.
- Os isoladores somente devem
assim, ser utilizados após a devida validação.
- A validação deve assim, considerar todos os
fatores críticos da tecnologia de isoladores; como por exemplo, a
qualidade do ar interna ao isolador e externa, a sanitização do isolador;
o processo de transferência de materiais e a integridade do isolador.
- O monitoramento deve ser
assim, realizado rotineiramente e deve também, incluir testes frequentes
de vazamento do isolador e do sistema de mangas/luvas.
A Instrução Normativa 35/2019 especifica a
tecnologia de sopro, envase e selagem.
- As unidades de sopro, envase
e selagem são equipamentos construídos para; em uma operação contínua,
formar recipientes a partir de granulados termoplásticos, envasar e selar,
tudo por uma máquina automática.
- O equipamento de sopro,
envase e selagem destinado à fabricação asséptica, equipado com sistema
eficaz de insuflamento de ar Grau A; pode ser assim, instalado em
um ambiente de no mínimo Grau C, desde que seja utilizado dessa
forma, vestimenta para grau A/B.
- O ambiente deve assim,
obedecer aos limites para partículas viáveis e não viáveis; em repouso e
apenas ao limite viável quando em operação.
- Os equipamentos de sopro,
envase e selagem utilizados para a fabricação de medicamentos
esterilizados terminalmente; devem ser assim, instalados em um ambiente de
no mínimo Grau D.
- Devido a essa tecnologia
especial, atenção particular deve ser assim, dada para com os seguintes
pontos:
- I -desenho e qualificação
de equipamentos;
- II -validação e
reprodutibilidade da limpeza no local (CIP) e da esterilização no local
(SIP);
- III -classificação de
limpeza da área onde o equipamento está instalado;
- IV -treinamento dos
operadores e paramentação;
- V -intervenções nas zonas
críticas do equipamento, incluindo qualquer montagem asséptica anterior
ao início do envase.
- Tecnologias e Processos.
Fonte:
antigo.anvisa.gov.br/documents
Monitoramento
e Controle
Monitoramento
e Controle de Equipamentos e Processos das áreas limpas.
A
Instrução Normativa 35/2019 especifica que as salas limpas e os equipamentos
que fornecem ar limpo devem ser monitorados rotineiramente em operação.
- Os pontos de amostragem para
monitoramento
devem ser assim, estabelecidos com base em um estudo formal de análise de
risco; e nos resultados obtidos durante a classificação das salas limpas
ou equipamentos que fornecem ar limpo.
- Para as áreas de Grau A,
o monitoramento de partículas deve ser assim, realizado ao longo de toda a
duração dos processos críticos; incluindo a montagem do equipamento,
exceto quando justificado pela presença de contaminantes no processo que
danificariam o contador de partículas; ou representariam um perigo, como
por exemplo organismos vivos e riscos radiológicos; onde nesses casos, o
monitoramento ao longo das operações de preparação do equipamento, antes
das situações impeditivas, deve ser assim, realizado.
- O monitoramento durante as
operações simuladas deve ser assim, executado.
- A área Grau A deve
ser assim, monitorada com tal frequência e tamanho de amostra adequados
que todas as intervenções; eventos transitórios e qualquer deterioração
do sistema possa ser dessa forma, capturada para que os limites de alerta
sejam então, disparados; caso excursões sejam detectadas.
- É aceitável que nem sempre
seja possível demonstrar baixos níveis de partículas ≥ 5,0 μm nos
processos de enchimento; quando este estiver em curso, devido à geração de
partículas ou gotículas do próprio produto.
Frequência de amostragem
- Um sistema similar de
monitoramento deve ser assim, utilizado para áreas Grau B; contudo,
a frequência de amostragem pode ser reduzida.
- A extensão do monitoramento
da área Grau B correlaciona-se com a efetividade da segregação
desta com a área Grau A que circunda.
- A área Grau B deve
ser assim, monitorada com tal frequência e tamanho de amostra adequados
que mudanças nos níveis de contaminação; ou qualquer deterioração do
sistema seja assim, capturada e os alarmes disparados caso os limites
sejam excedidos.
- Os sistemas de monitoramento
de partículas podem consistir de contadores de partículas independentes;
uma rede de pontos de amostragem conectados por um tubo de distribuição a
um único contador de partículas ou uma combinação de ambos.
- O sistema selecionado deve
ser assim, apropriado para o tamanho das partículas consideradas.
- Quando sistemas de
amostragem remota forem usados, o comprimento da tubulação e os raios de
quaisquer curvas na tubulação; devem ser assim, considerados no contexto
de perdas de partículas na tubulação.
- A seleção do sistema de
monitoramento deve considerar qualquer risco apresentado pelos materiais
utilizados na fabricação, por exemplo, organismos vivos ou radiofármacos.
- Os tamanhos das amostras
tomadas para fins de monitoramento por sistemas automatizados normalmente
serão uma função da taxa de amostragem do sistema usado.
- Não é necessário que o
volume da amostragem seja o mesmo usado para a classificação das salas
limpas e dos equipamentos que fornecem ar limpo.
Monitoramento da concentração
- Nas áreas de Grau A e B,
o monitoramento da concentração de partículas ≥5,0 μm assume um
significado particular, pois é uma importante ferramenta de diagnósticos
na detecção antecipada de falhas.
- A indicação ocasional de
contagem de partículas ≥ 5,0 μm pode ocorrer por contagens falsas
ocasionadas por ruído eletrônico, luzes difusas, coincidências etc.
- No entanto, a contagem
consecutiva ou regular de níveis baixos é um indicador de um possível
evento de contaminação e deve ser asim, investigada.
- Tais eventos podem indicar
falhas precoces do sistema HVAC, falhas no equipamento de envase; podendo
também diagnosticar práticas inadequadas durante a configuração da
máquina e a operação de rotina.
- Os limites de partículas
dados na tabela do art. 10 para o estado “em repouso” devem ser assim,
alcançados após um curto período de limpeza de 15 a 20 minutos (valor
orientativo); sem pessoal em um estado automatizado, após a conclusão das
operações.
- O monitoramento das áreas de
Grau C e D; em operação deve ser assim, realizado de acordo com os
princípios de gerenciamento de riscos na qualidade.
- Os requerimentos e limites
de alertas/ação dependem da natureza das operações realizadas, mas o tempo
de recuperação requerido deve ser assim, alcançado.
- O monitoramento de
temperatura e umidade relativa, assim como outros parâmetros, dependem do
produto e da natureza das operações realizadas.
- Estes parâmetros não devem
assim, interferir no grau de limpeza definido para a área.
As tabelas abaixo exemplificam as operações que
podem ser assim, realizadas nos diferentes graus de limpeza. Monitoramento e
Controle.
 |
| Classificação Sistema sala limpa |
- Quando da realização de
operações assépticas, o monitoramento deve ser frequente, por meio de
métodos como placas de sedimentação; amostragem de ar volumétrica e de
superfície (por exemplo:
cotonetes swab e placas de contato). (Retificado
no DOU nº 49, de 12 de março de 2020)
- Os métodos de amostragem
usados nestas operações não podem interferir na proteção conferida ao
medicamento pela área limpa.
- Superfícies e pessoal devem
ser assim, monitorados após operações críticas.
- Os resultados do
monitoramento devem ser assim, considerados ao se revisar a documentação
do lote para a liberação do produto acabado.
- Monitoramento microbiológico
adicional pode ser assim, requerido fora das situações de fabricação, como
por exemplo depois da validação de sistemas, limpeza e sanitização.
Os limites recomendados para o monitoramento
microbiológico de áreas limpas durante operação de monitoramento e controle,
estão dados na tabela abaixo.
 |
Classificaça do Sistema sala limpa
|
- Devem ser assim,
estabelecidos limites adequados de alerta e ação para os resultados do
monitoramento microbiológico e de partículas.
- Se esses limites forem
excedidos, os procedimentos operacionais devem assim, descrever as ações
corretivas.
- u/placa) Luva 5 dedos (cfu/luva)1
A < 1 < 1 < 1 < 1 B 10 5 5 5 C 100 50 25 - D 200 100 50 - 1 –
O limite disposto é a especificação para a luva direita bem como a
esquerda, sendo necessário na rotina o teste de ambas as luvas do operador
- Devem ser assim,
estabelecidos limites adequados de alerta e ação para os resultados do
monitoramento microbiológico e de partículas.
- Se esses limites forem
excedidos, os procedimentos operacionais devem assim, descrever as ações
corretivas.
Fonte:
antigo.anvisa.gov.br/documents
Outros Canais Importantes:
Código Sanitário do Estado de São Paulo
Código Sanitário do Município de São Paulo
Sistema Estadual de Saúde do Estado do Amazonas
Estado de Alagoas
Estado de Goiás
Sistema Estadual de Vigilância Sanitária da Paraíba
Política estadual de resíduos sólidos do Estado do Ceará
Sistema de Saúde do Estado do Piauí
Serviços de saúde no Estado do Paraná
 |
Divisória para Sala Limpa
|



















{kind=link}